Ten Injuries and One Death Lead to Medtronic Brain Stent Recall
Editors carefully fact-check all Drugwatch.com content for accuracy and quality.
Drugwatch.com has a stringent fact-checking process. It starts with our strict sourcing guidelines.
We only gather information from credible sources. This includes peer-reviewed medical journals, reputable media outlets, government reports, court records and interviews with qualified experts.
In February 2020, Medtronic recalled 1,812 units of its Pipeline Flex Embolization Device and Pipeline Flex Embolization Device with Shield Technology after it found the device’s delivery system could fracture during implantation.
In March, the U.S. Food and Drug Administration classified it as a Class I recall — the agency’s most serious recall for products that could cause serious harm or death. Of the products recalled globally, 822 were sold in the United States.
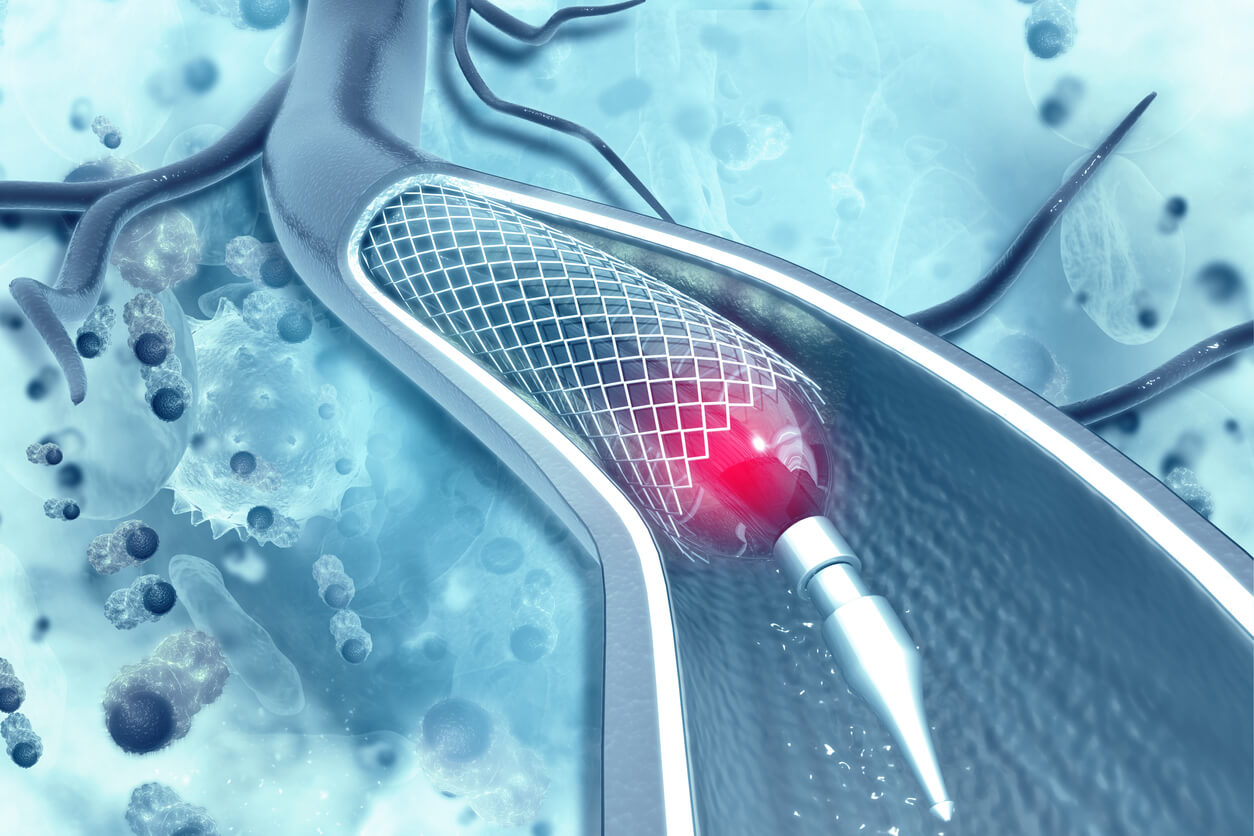
Doctors use the embolization devices to treat brain aneurysms, bulges or sacs in blood vessels in the brain. The aneurysms can leak or rupture and cause life-threatening brain bleeds.
Pipeline Flex Embolization Devices are cylinder-shaped, permanent mesh implants made of braided platinum, tungsten and cobalt-chromium-nickel alloy. Surgeons insert the devices with a guidewire delivery system into the body through a blood vessel in the leg and guide it to the site of the aneurysm. Doctors then release the stent, and it restricts blood from entering the bulge of the aneurysm.
Medtronic recalled the devices because the delivery system could fracture, leaving the broken pieces in a patient’s brain. These pieces could then travel in the brain bloodstream.
Complications from Device Fractures
Medtronic received 50 reports with 10 injuries and one death linked to the recalled devices from November 2019 to March 2020.
Leaving fractured pieces in the body or attempting to remove them can worsen a patient’s condition. It can also cause other serious complications including stroke, blood vessel blockage and death.
A search of the FDA’s Manufacturer and User Facility Device Experience (MAUDE) database shows about 500 adverse event reports related to the Pipeline Flex from May 8, 2019 to May 22, 2020. Because these reports are voluntary, the FDA cannot verify them.
The company sent an Urgent Medical Device Recall Notice to their customers on Feb. 14, 2020. The letter advised customers to remove all affected products from their inventory and not to use them. Patients who already had the devices implanted don’t have any increased health risks from the recalled devices.
Medtronic previously recalled its Pipeline Classic and Pipeline Flex in 2013, 2014, 2015 and 2016 for labeling problems, device malfunctions and a defective device coating that could flake off into the blood stream.