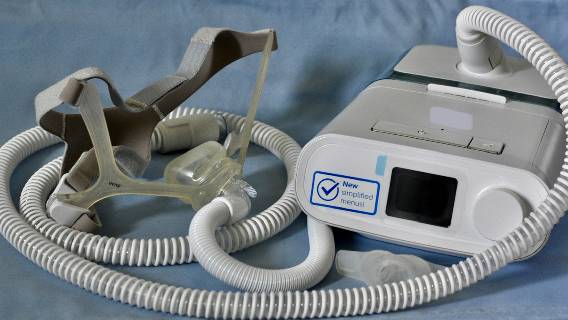

Philips Respironics has recalled another wave of its DreamStation CPAP and BiPAP machines over concerns they could fail to deliver therapy to patients.
“The FDA has identified this as a Class I recall, the most serious type of recall. Use of these devices may cause serious injuries or death,” according to the U.S. Food and Drug Administration news alert issued April 7.
Approximately 1,200 remediated DreamStation devices were recalled by the company in February because they were incorrectly programmed. During remediation, the devices were erroneously given either an incorrect serial number or a duplicate serial number, which could result in improper therapy, or no therapy at all, for the user.
“As a result, patients may find the device provides different pressure and comfort settings than the patient is accustomed to,” Philips spokesperson Steve Klink wrote in an email to DrugWatch.com.
There is no warning or indication to the user that the machine is defective. Incorrect therapy or therapy failure may lead to several health conditions, including respiratory failure, heart failure, serious injury or death, according to the FDA notice.
Users can continue to use the impacted devices but should contact the company for a new Philips CPAP device, Klink said.
There have been 43 complaints about the current recall issue, but no reports of injury or death, according to the FDA.
“Philips Respironics is reaching out to patients to set up replacement and return of affected devices. No further action is necessary by patients. To date, we are more than halfway [through] with the shipments of replacement devices to patients,” Klink wrote.
Latest Action Adds to Millions of Philips CPAP Recalls
In 2021, Philips recalled millions of its first-generation DreamStation CPAP machines because degraded polyester-based polyurethane foam inside the machines meant to reduce noise was breaking off and blowing into the user’s mouth. Philips warned that ingesting or inhaling particles or gases from broken-down PE-PUR foam could cause toxic respiratory and carcinogenic effects.
As of Dec. 31, 2022, the FDA had received more than 98,000 medical device reports of side effects linked to PE-PUR foam. Reported side effects include cough, dizziness and headache, as well as serious adverse effects such as asthma, breathing problems, cancer, chest pain, infection, nodules, pneumonia and respiratory problems.
Cancers linked to Philips CPAP include bladder cancer, stomach cancer, lung cancer and kidney cancer. There have also been reports of 346 related deaths, according to the FDA.
People who used Philips’ recalled CPAP machines have filed Philips CPAP lawsuits after suffering injuries. These lawsuits claim the Philips CPAP devices are defective and the company did not adequately warn about the risk of serious injury.
Current Devices Recalled by Philips Respironics
CPAP and BiPAP devices are used to help people with breathing conditions continue to breathe at a regular rhythm. Often people with sleep apnea will use a CPAP or BiPAP device while sleeping to decrease episodes.
CPAP machines work by delivering pressurized air through a tube to a mask that covers the nose and mouth or just the nose. A BiPAP machine pumps air into the lungs. It uses higher pressure when you breathe in and lower pressure when you breathe out.
Devices included in the recall are:
Product Name: Philips Respironics DreamStation1 (Uno Remediated Devices)
Product Models:
- DOM-RECRT
- DreamStation Auto
- FR REP DreamStation Auto BiPAP
- REP DreamStation Auto CPAP Recert
- REP DreamStation Auto CPAP, DOM – RECRT
Serial Numbers: See Medical Device Recall Database
Distribution Dates: Dec. 1, 2021 to Oct. 31, 2022
The devices could be used in health care settings and by users at home. Users can check the device’s serial number to find out if it is part of the recall.
Health care professionals and consumers may report adverse reactions or quality problems they experienced using these devices to the FDA’s MedWatch reporting program using the online form or they can call 1-800-332-1088 for more information.