Deaths Linked to Philips CPAP, BiPAP Machines Rising
Editors carefully fact-check all Drugwatch.com content for accuracy and quality.
Drugwatch.com has a stringent fact-checking process. It starts with our strict sourcing guidelines.
We only gather information from credible sources. This includes peer-reviewed medical journals, reputable media outlets, government reports, court records and interviews with qualified experts.
A new report released by the U.S. Food and Drug Administration shows the number of patient deaths connected to the use of recalled Philips Respironics CPAP, BiPAP and ventilators continues to rise. An additional 40 deaths were reported to the FDA in the first quarter of 2023, bringing the total number of patient deaths to 385.
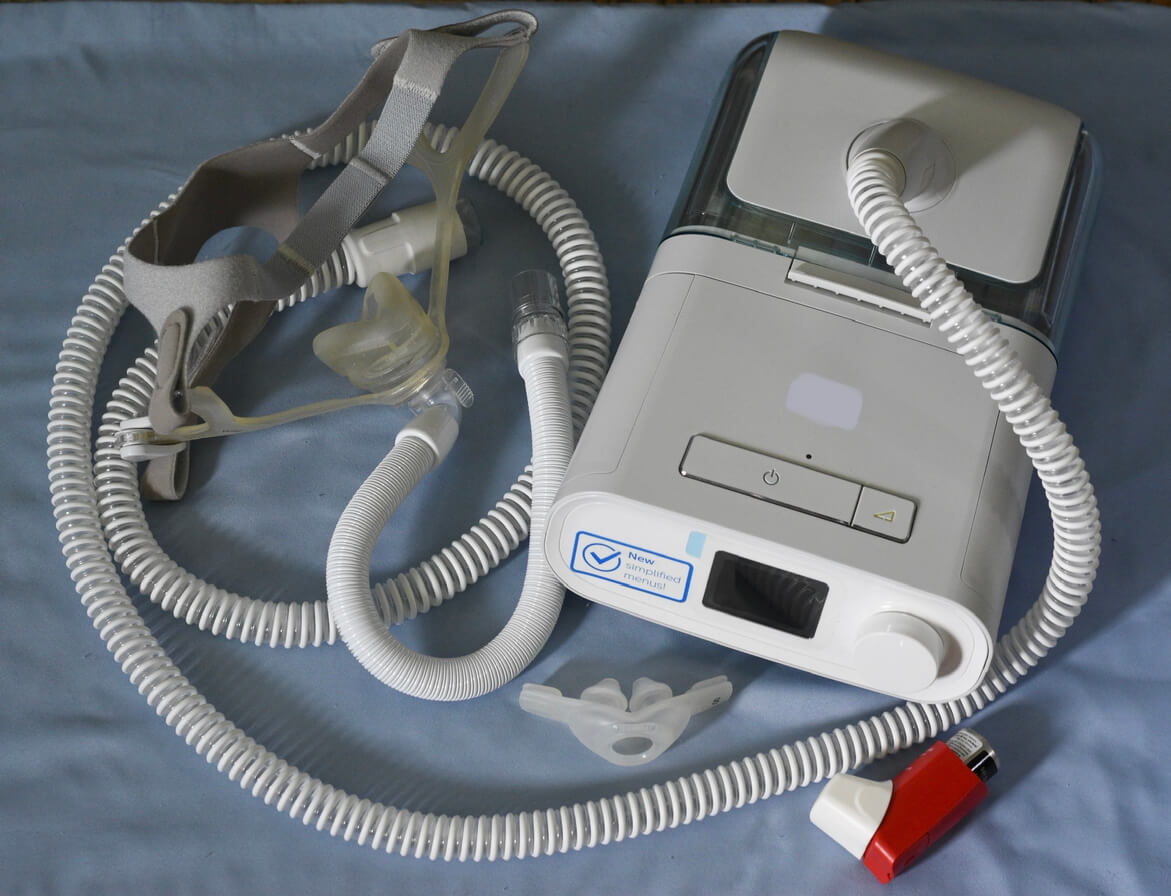
Polyester-based polyurethane (PE-PUR) foam used inside the devices to reduce sound and vibration can break down and be breathed in or swallowed by the patient using the device. That could cause issues and injuries to the patient that range from irritation of the skin to cancer or even death.
Philips is the largest maker of continuous positive airway pressure machines in the world. Philips CPAP machines are used to treat obstructive sleep apnea and may have benefits for other health conditions as well. CPAP side effects are possible.
Philips CPAP Recalls Follow Injury Reports
In June 2021, Philips recalled certain ventilators, BiPAP machines and CPAP machines after reports of patient injuries because of the deteriorating foam inside the machines.
Shortly after the 2021 recall was announced, lawyers began investigating Philips CPAP lawsuits on behalf of patients who used recalled devices and were diagnosed with cancer or other health issues such as respiratory problems.
Certain Trilogy Evo Ventilators with specific serial numbers, distributed from April 15, 2021 to May 24, 2021, were also recalled. A list of the devices that Philips recalled between 2009 and April 26, 2021, can be found in the FDA safety communication published June 2.
Medical Device Reports Track Adverse Events
During the first quarter of 2023, the FDA received more than 6,000 medical device reports, the agency wrote in its June 2 safety communication.
Manufacturers such as Philips are required to submit MDRs when they become aware of an event “that reasonably suggests that one of their devices may have caused or contributed to a death or serious injury, or has malfunctioned and that device or a similar device marketed by the manufacturer would be likely to cause or contribute to a death or serious injury if the malfunction were to recur,” the FDA stated.
People can also voluntarily submit reports of adverse events and malfunctions to the FDA. Since April 2021, the agency said it has received more than 105,000 MDRs, which include 385 reports of death reportedly connected to the PE-PUR foam breakdown or suspected foam breakdown. These include:
- April 1, 2021-April 30, 2022: More than 21,000 MDRs and 123 deaths.
- May 1-July 31, 2022: More than 48,000 MDRs and 45 deaths.
- Aug. 1-Oct. 31, 2022: More than 21,000 MDRs and 96 deaths.
- Nov. 1-Dec. 31, 2022: More than 8,000 MDRs and 81 deaths.
- Jan. 1-March 31, 2023: More than 6,000 MDRs and 40 deaths.
The number of deaths between November and December 2022 increased to 81 deaths after Philips’ retrospective review of MDRs, the FDA said.
“Although MDRs are a valuable source of information, this passive surveillance system has limitations. The incidence, prevalence, or cause of an event cannot typically be determined from this reporting system alone due to under-reporting of events, inaccuracies in reports, lack of verification that the device caused the reported event, and lack of information about details such as frequency of device use,” the FDA report stated.
The FDA said it continues to review MDRs and will release new information as it becomes available.